tutorial_zmatrix
Using Z-matrices
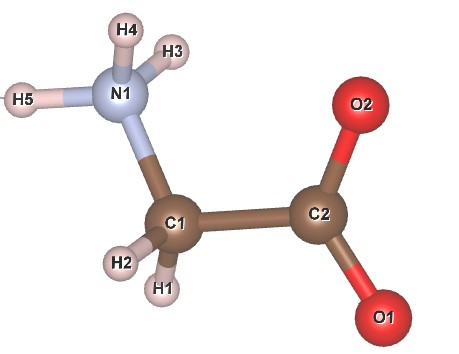
Learning outcome: Z-matrices can be a convenient way of describing molecules or clusters for either structure solution or structure refinement. Instead of refining on atomic coordinates you refine the molecule’s position and rotation in space, plus internal degrees of freedom such as torsion angles or bond lengths. This tutorial uses a simulated data set from the zwitter ionic form of glycine to explore these ideas. The structural model is from an 18 K neutron analysis by Aree and Burgi. A labelled sketch of the molecule is:
Files needed: glycine_cuka1_01.xye; glycine_865358.cif; glycine_Zwitter.gzmat (save without .txt extension)
An INP file that contains the syntax used in the tutorial is here.
A simple Rietveld with a prewritten Z-matrix
1. Set up to do a simple Rietveld refinement using the cif file glycine_865358.cif as your starting model. Assume the data were recorded on a diffractometer with Cu Ka1 radiation and a Ge 111 monochromator (angle 27.26 degrees). Fit the data from 5 to 90 degrees. Use a single equated isotropic temperature factor for C/N/O and a different equated one for all H. You should be able to get an Rwp of ~3.96 %.
2. The text below contains a Z-matrix description of glycine in TOPAS syntax. Check that it is consistent with the molecular sketch above. Introduce this to your INP file and repeat the refinement. The text needs to go in the str section of the file, and the site labels in the Z-matrix description must match those in the sites lines. You should get Rwp ~26 %.
rigid
z_matrix N1
z_matrix C1 N1 1.48170
z_matrix H3 N1 1.03915 C1 109.4912
z_matrix H4 N1 1.03915 C1 109.4912 H3 240.0000
z_matrix H5 N1 1.03915 C1 109.4912 H3 120.0000
z_matrix C2 C1 1.50913 N1 109.4912 H3 60.0000
z_matrix H1 C1 1.17186 N1 109.4912 H4 180.0000
z_matrix H2 C1 1.17186 N1 109.4912 H3 180.0000
z_matrix O1 C2 1.27987 C1 120.0000 N1 180.0000
z_matrix O2 C2 1.27987 C1 120.0000 O1 180.0000
macro rotcon {val_on_continue = Rand(-180,180);}
Rotate_about_axies(!xrot -60 rotcon, !yrot 0 rotcon, !zrot 70 rotcon )
Translate(!xtrans 0.30 , !ytrans 0.1 , !ztrans 0.75)3. Try refining the rigid body translations and rotation angles (remove the ! signs in the corresponding macros). You should get Rwp ~8.3 %.
4. If you look at your atomic sites in topas-editor you’ll see that the coordinates are listed in red showing they have been refined. This happens without you having to introduce @ symbols or parameter names as the coordinates are replaced by those calculated from the rigid body.
5. Try refining the torsion angle that describes the rotation of the CO2– group around the C1-C2 bond. You should get Rwp ~3.85 %. You can do this by putting the @ symbol after N1 on the last but one line of the z_matrix description:
z_matrix O1 C2 =d_CO;:1.27987 C1 120.0000 N1 @ -1806. You could try refining the bond distances and angles contained in the Z-matrix. One way of doing that is below.
prm d_CN 1.48051747 min 1 max 1.6
prm d_NH 1.04262098 min 1 max 1.6
prm d_CC 1.50894262 min 1 max 1.6
prm d_CH 1.16277196 min 1 max 1.6
prm d_CO 1.27940825 min 1 max 1.6
rigid
z_matrix N1
z_matrix C1 N1 =d_CN;:1.48052
z_matrix H3 N1 =d_NH;:1.04262 C1 109.4912
z_matrix H4 N1 =d_NH;:1.04262 C1 109.4912 H3 240.0000
z_matrix H5 N1 =d_NH;:1.04262 C1 109.4912 H3 120.0000
z_matrix C2 C1 =d_CC;:1.50894 N1 109.4912 H3 60.0000
z_matrix H1 C1 =d_CH;:1.16277 N1 109.4912 H4 180.0000
z_matrix H2 C1 =d_CH;:1.16277 N1 109.4912 H3 180.0000
z_matrix O1 C2 =d_CO;:1.27941 C1 120.0000 N1 @ -160.68239
z_matrix O2 C2 =d_CO;:1.27941 C1 120.0000 O1 180.0000
macro rotcon {val_on_continue = Rand(-180,180);}
Rotate_about_axies(xrota -61.93217 rotcon, yrota -1.42539 rotcon, zrota 68.81819 rotcon )
Translate(xtransa 0.29672 , ytransa 0.08694 , ztransa 0.74246)How to match your Z-matrix to known coordinates
1. One common use of Z-matrix descriptions is when you have a partial structural solution (e.g. from simulated annealing or charge flipping) but want to replace the approximate coordinates with a Z-matrix description. This can be done by “matching” the coordinates of your rigid body to the partial coordinates using an “only_penalties” refinement that tries to minimise the distances between the reference coordinates and those calculated for the rigid body.
2. Create a list of reference coordinates by copy/pasting the ideal coordinates. Rename each site with _m for “match”. Change the occupany of each site to zero so the atoms don’t contribute in any later Rietveld fit. Introduce lines to match up individual sites and the corresponding macro. Set the rigid body translations and rotations to 0 initially.
At the top of the INP file add the lines:
#define match_sites
continue_after_convergence
iters 1000In the rigid body change the translations and angles to:
Rotate_about_axies(xrota 0 rotcon, yrota 0 rotcon, zrota 0 rotcon )
Translate(xtransa 0 , ytransa 0 , ztransa 0)In the str section (e.g. just below the rigid body) add the lines:
#ifdef match_sites
only_penalties
'ideal coordinates from single crystal structure to match to
site N1_m x 0.29501 y 0.08830 z -0.25953 occ N 0
site H3_m x 0.2923 y 0.00181 z -0.2258 occ H 0
site H4_m x 0.4916 y 0.11821 z -0.1309 occ H 0
site H5_m x 0.2839 y 0.09861 z -0.4553 occ H 0
site C1_m x 0.05795 y 0.14585 z -0.21433 occ C 0
site H1_m x -0.1437 y 0.11556 z -0.3590 occ H 0
site H2_m x 0.0735 y 0.23727 z -0.2423 occ H 0
site C2_m x 0.06920 y 0.12527 z 0.06578 occ C 0
site O1_m x -0.15495 y 0.14232 z 0.10651 occ O 0
site O2_m x 0.30223 y 0.09361 z 0.23646 occ O 0
'the 4 in the macro is the number of molecules in the cell, this means final value in macro is distance between atoms
Match_Site(N1_m, N*, 1, , 4, 0.0221326416)
Match_Site(H3_m, H*, 1, , 4, 0.0296391439)
Match_Site(H4_m, H*, 1, , 4, 0.0624116228)
Match_Site(H5_m, H*, 1, , 4, 0.0444855656)
Match_Site(C1_m, C*, 1, , 4, 0.0337141799)
Match_Site(H1_m, H*, 1, , 4, 0.0344161913)
Match_Site(H2_m, H*, 1, , 4, 0.0340206987)
Match_Site(C2_m, C*, 1, , 4, 0.0273388281)
Match_Site(O1_m, O*, 1, , 4, 0.0345707072)
Match_Site(O2_m, O*, 1, , 4, 0.00959288178)
macro Match_Site(s1, s2, wby, c, Z, Rcalc)
{
#m_ifarg c ""
#m_unique_not_refine c
#m_endif
box_interaction to_N 0 s1 s2 c = R;
local =c/Z;: Rcalc
penalty = (wby) c^2;
}
#endif3. Run the file for a few thousand iterations then stop it. You should get chi2 about 0.47. If you look at the Match_Site macro All the atoms should be within about 0.02 A of their ideal positions.
4. Comment out the #define match_sites. The Rietveld should converge rapidly to 3.85%.
5. This example matched all the atoms. In practice you might match a large Z-matrix to just a few sites.
Creating a Z-matrix from scratch
This part of the tutorial discusses different ways to create a Z-matrix from scratch.
1. If you’re mega-keen try working it out yourself. The syntax is in the TOPAS techref or Chapter 6 of the textbook by Dinnebier, Leineweber and Evans. It’s good to do this once for a simple molecule. If you’re mega-lazy and mega-optimistic, ChatGPT can sometimes make sensible suggestions.
2. If you have a structure that already contains the molecule you’re interested in you can use view_structure in TOPAS to visualise the structure. In this case we could use the structure in our original Rietveld INP file. In other cases you could just simulate a pattern from a known cif. In the structure viewer click on the “box/tick” icon in the top left. In the options menu click on “First guess Z matrix”. The temporary output window will list various possible Z-matrices for molecules or fragments found in the unit cell. In this example the third one down should describe a complete glycine molecule.
3. The Bruker version of TOPAS has a built-in rigid body editor.
4. If you have a .mol file containing your coordinates you can use open babel to convert it to a .gzmat file. This is a Gaussian format z-matrix. Instructions on installing open babel are below. You’ll need a command like:
./obabel.exe glycine.mol -O glycine.gzmat5. You can produce a .mol file in various packages. One option is to sketch a molecule in mercury, let it do the 2D to 3D conversion, then export as a .mol.
6. For the glycine example in this tutorial you can create the Z-matrix of the zwitterionic form directly from a smiles string in open babel. The command needed is:
.\obabel.exe -:"[NH3+]CC(=O)(O-)" --gen3d -O glycine_Zwitter.gzmat7. open babel automatically adds H atoms with the above command. In this case it adds a H to the O- species. This can be removed manually from the final TOPAS Z-matrix file. The C-O bond lengths should also be equated to reflect their true delocalised nature. Remember that you’re trusting open babel’s default optimiser to create the bond distances and angles. You will probably want to adjust these.
8. You can create a smiles string using a variety of different tools. One is the web editor at: https://www.bondcraft.net/. Sketch your molecule then click “copy smiles” in the top right. ChatGPT might also work.
9. You should be able to read the .gzmat directly using topas-editor. Download the .gzmat here and save without the .txt extension. Go to “miscellaneous useful commands” and select “Insert Z-matrix from Gaussian .gzmat”. This should automatically create a series of site lines with the same labels as the Z-matrix. You should delete any pre-existing site lines. Repeat the Rietveld refinement. Investigate the impact of changing bond lengths, bond angles and torsion angles.
10. If the topas-editor method doesn’t work, the Gaussian format .gzmat file can be converted to TOPAS format with the convert.py script by Marko Vardo on github. Note that Marko used Avogardro to produce .gzmat files, I couldn’t get a free version to work.
11. If neither of these methods works, the text below contains the obabel-derived Z-matrix converted to TOPAS format. Note that you’ll need to generate matching sites by hand.
rigid
load z_matrix {
N1
C2 N1 1.5035
C3 C2 1.5175 N1 109.72
O4 C3 1.2567 C2 120.16 N1 tor1 19.32112
O5 C3 1.2567 C2 116.60 N1 =180+tor1;
H6 N1 1.0253 C2 114.27 C3 180.00
H7 N1 1.0318 C2 109.72 C3 56.94
H8 N1 1.0318 C2 109.72 C3 303.07
H9 C2 1.0921 N1 106.73 C3 240.06
H10 C2 1.0921 N1 106.74 C3 119.93
}
macro rotcon {val_on_continue = Rand(-180,180);}
Rotate_about_axies(xrota 12.56321 rotcon, yrota -83.01635 rotcon, zrota -45.86182 rotcon )
Translate(xtransa 0.29197 , ytransa 0.08887 , ztransa 0.73657)Structure solution
This same INP file can be used for structure solution. Set the rigid body rotations and translations to zero. Turn on continue_after_convergence. The simple randomisation of molecular rotations is sufficient that the structure can be solved in a few thousand iterations.
Other examples
The organic Rietveld tutorial explores using restraints, conventional rigid bodies and a Z-matrix approach.
Installing Open Babel
open babel is a free tool for manipulating lots of different file formats and can produce Z-matrices automatically. For me (February 2026) the default installation from the downloadable .exe didn’t work perfectly out of the box. I had to:
1. First install from the .exe file available on github here.
2. Download the full .zip file, extract, then manually copy all the files in the “data” folder of the .zip into the main open babel folder (C:\Program Files\OpenBabel-3.1.1).
3. I had to run obabel directly from a command prompt in this folder for the 3D coordinate generation to work (data files not found otherwise).
4. This is probably due to environment variables not being set properly on my managed PC.
5. I used commands like:
C:\Program Files\OpenBabel-3.1.1>.\obabel.exe -:"[NH3+]CC(=O)(O-)" --gen3d -O glycine_Zwitter.gzmat