Tutorial organicriet
Organic Rietveld refinement
Files needed: d8_00796.raw; rigidrod.cif; rod.gzmat (save without .txt extension)
Learning Outcomes: This example shows a Rietveld refinement of an organic molecule using (relatively low quality) lab data. The use of restraints, rigid bodies and Z-matrices is illustrated. Warning: some of the concepts here are relatively advanced! The molecule being used is:
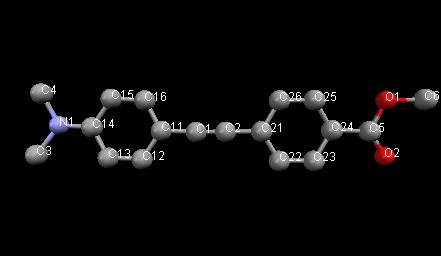
Topas advantages: lots of different ways of using restraints, rigid bodies, etc.
Restrained refinement
1. Save the datafile and cif file to your working directory.
2. Work through the menus in the TOPAS_Durham/Rietveld refinement section of topas-editor or jEdit.
3. Click on “Select Data File” and navigate to find the file d8_00796.raw.
4. In “2. Instrument” select “BB_CuKa1_Ge111_PSD Durham” – the diffractometer used to collect the data.
5. In “3. Corrections” click on “Refine zero point” to flag that zero should be refined.
6. In “4. Rietveld or Pawley” select “Structure from CIF” and read in rigidrod.cif.
7. Set up to refine a single temperature factor on each atom (add “beq bvalue 1” at the end of each site line. Set to refine a, b and c cell parameters. Set to refine from 4 to 50 degrees (add lines saying “start_X 4” and “finish_X 50” after the xdd filename line.
8. You should be able to get Rwp=11.74%.
9. Before refining atomic coordinates it’s sensible to introduce restraints to prevent atoms refining to silly positions. Those listed below restrain benzene rings to be regular (via distances and angles) and planar (via the flatten restraint) and the NMe2 group on the molecule to be planar. Each macro contains the sites to be restrained, the ideal distance, the actual distance, a tolerance (difference before restraint applied) and a weight for the restraint.
'restrain one benzene ring
Distance_Restrain(C11 C12, 1.400, 1.43463, 0, 1)
Distance_Restrain(C12 C13, 1.400, 1.41595, 0, 1)
Distance_Restrain(C13 C14, 1.400, 1.60138, 0, 1)
Distance_Restrain(C14 C15, 1.400, 1.45696, 0, 1)
Distance_Restrain(C15 C16, 1.400, 1.40217, 0, 1)
Distance_Restrain(C16 C11, 1.400, 1.49306, 0, 1)Angle_Restrain(C11 C12 C13,120, 118.52024,0, .001)
Angle_Restrain(C12 C13 C14,120, 118.41604,0, .001)
Angle_Restrain(C13 C14 C15,120, 118.54301,0, .001)
Angle_Restrain(C14 C15 C16,120, 120.52919,0, .001)
Angle_Restrain(C15 C16 C11,120, 119.61364,0, .001)
Angle_Restrain(C16 C11 C12,120, 123.25791,0, .001)Flatten(C11 C12 C13 C14 C15 C16,0, 0.0188811239,10)'restrain second benzene ring
Distance_Restrain(C21 C22, 1.400, 1.42104, 0, 1)
Distance_Restrain(C22 C23, 1.400, 1.37979, 0, 1)
Distance_Restrain(C23 C24, 1.400, 1.43837, 0, 1)
Distance_Restrain(C24 C25, 1.400, 1.39612, 0, 1)
Distance_Restrain(C25 C26, 1.400, 1.48327, 0, 1)
Distance_Restrain(C26 C21, 1.400, 1.38858, 0, 1)Angle_Restrain(C21 C22 C23,120, 120.62386,0, .001)
Angle_Restrain(C22 C23 C24,120, 120.88302,0, .001)
Angle_Restrain(C23 C24 C25,120, 119.45380,0, .001)
Angle_Restrain(C24 C25 C26,120, 118.98380,0, .001)
Angle_Restrain(C25 C26 C21,120, 119.40741,0, .001)
Angle_Restrain(C26 C21 C22,120, 120.31336,0, .001)Flatten(C21 C22 C23 C24 C25 C26,0, 0.0072359109,10)'restrain central linking group
Distance_Restrain(C21 C2 , 1.430, 1.60025, 0, 1)
Distance_Restrain(C11 C1 , 1.430, 1.40512, 0, 1)
Distance_Restrain(C2 C1 , 1.200, 1.27122, 0, 1)'restrain nme2 group
Distance_Restrain(C14 N1 , 1.380, 1.54348, 0, 1)
Distance_Restrain(N1 C3 , 1.450, 1.45761, 0, 1)
Distance_Restrain(N1 C4 , 1.450, 1.49031, 0, 1)
Flatten(N1 C3 C4 C14,0, 0.000726450936,10)'restrain CO2CH3 group
Distance_Restrain(C24 C5 , 1.500, 1.58680, 0, 1)
Distance_Restrain(C5 O1 , 1.350, 1.38465, 0, 1)
Distance_Restrain(C5 O2 , 1.200, 1.25119, 0, 1)
Distance_Restrain(O1 C6 , 1.440, 1.50432, 0, 1)10. Flag all atomic coordinates to refine. You should get Rwp of 7.99%. Have a look at the ideal and restrained bond distances in the .inp file after refinement. Try using “view_structure” in your input file to look at the molecule. In case you mess up an example file is here.
Rigid body refinement
1. We can try the same refinement using rigid bodies to describe the benzene rings. This can be done by introducing the lines below immediately below the last atomic coordinate line in the file:
rigid
Hexagon_sitting_on_side_in_xy_plane(C12, C13, C16, C15, C11, C14, 1.4)
Rotate_about_axies( @ 55.30755`, @ 117.44639`, @ 356.92214`)
Translate( @ 0.63680`, @ 0.48817`, @ 0.58321`)rigid
Hexagon_sitting_on_side_in_xy_plane(C22, C23, C26, C25, C21, C24, 1.4)
Rotate_about_axies( @ 112.97294`, @ 244.56505`,@ -176.67930`)
Translate( @ 0.15133`, @ 0.50553`, @ 0.38959`)The coordinates for atoms in benzene rings are now replaced by those of an ideal hexagon whose position and rotation is refined. Because atomic sites are no longer refined independently you must remove the “@” symbols from the coordinate lines of C11-C26. i.e. the file should look like:
site N1 x @ 0.85749` y @ 0.54252` z @ 0.65583` occ N 1 beq bvalue 3.0675`
site O1 x @ -0.29459` y @ 0.42989` z @ 0.30397` occ O 1 beq bvalue 3.0675`
site C1 x @ 0.43507` y @ 0.48261` z @ 0.50899` occ C 1 beq bvalue 3.0675`
site C2 x @ 0.37059` y @ 0.52090` z @ 0.47274` occ C 1 beq bvalue 3.0675`
site O2 x @ 0.03491` y @ 0.54861` z @ 0.28399` occ O 1 beq bvalue 3.0675`
site C3 x @ 1.02060` y @ 0.62899` z @ 0.67088` occ C 1 beq bvalue 3.0675`
site C4 x @ 0.70753` y @ 0.44862` z @ 0.68755` occ C 1 beq bvalue 3.0675`
site C5 x @ -0.07888` y @ 0.47581` z @ 0.31011` occ C 1 beq bvalue 3.0675`
site C6 x @ -0.39808` y @ 0.47302` z @ 0.26452` occ C 1 beq bvalue 3.0675`
site C11 x 0.53155 y 0.49281 z 0.54557 occ C 1 beq bvalue 3.0675`
site C12 x 0.73453 y 0.57636 z 0.55047 occ C 1 beq bvalue 3.0675`
site C13 x 0.83977 y 0.57172 z 0.58811 occ C 1 beq bvalue 3.0675`
site C14 x 0.74205 y 0.48354 z 0.62085 occ C 1 beq bvalue 3.0675`
site C15 x 0.53908 y 0.39999 z 0.61595 occ C 1 beq bvalue 3.0675`
site C16 x 0.43383 y 0.40463 z 0.57831 occ C 1 beq bvalue 3.0675`
site C21 x 0.24937 y 0.51019 z 0.42789 occ C 1 beq bvalue 3.0675`
site C22 x 0.36026 y 0.57890 z 0.39422 occ C 1 beq bvalue 3.0675`
site C23 x 0.26221 y 0.57423 z 0.35591 occ C 1 beq bvalue 3.0675`
site C24 x 0.05328 y 0.50086 z 0.35129 occ C 1 beq bvalue 3.0675`
site C25 x -0.05761 y 0.43216 z 0.38496 occ C 1 beq bvalue 3.0675`
site C26 x 0.04044 y 0.43682 z 0.42327 occ C 1 beq bvalue 3.0675`rigid
Hexagon_sitting_on_side_in_xy_plane(C12, C13, C16, C15, C11, C14, 1.4)
Rotate_about_axies( @ 55.30755`, @ 117.44639`, @ 356.92214`)
Translate( @ 0.63680`, @ 0.48817`, @ 0.58321`)rigid
Hexagon_sitting_on_side_in_xy_plane(C22, C23, C26, C25, C21, C24, 1.4)
Rotate_about_axies( @ 112.97294`, @ 244.56505`,@ -176.67930`)
Translate( @ 0.15133`, @ 0.50553`, @ 0.38959`)When you refine you should be able to get Rwp=6.1% after adding e.g. 6 more background terms. Note that in the output file the restraints for the rings are now “perfect”. They’re redundant and could be deleted. In case you mess up, look at the file linked here.
Refinement using a Z-matrix
In the above exercises we started from a known model. We can also work with a Z-matrix built without structural knowledge to both refine and solve the structure. It might help if you’ve done the tutorial on Z-matrices first.
1. Start from the INP file you prepared above. You might choose to fix paramers like peak shape, unit cell, zero, etc.
2. Delete all the site information and any restraints or rigid bodies in the file.
3. You should be able to read the Z-matrix directly from rod.gzmat using “Miscellaneous useful commands/Insert Z-matrix from Gaussian .gzmat”. If that doesn’t work, paste in the Z-matrix information below. The .gzmat was created as described in the Z-matrix tutorial using open babel. The open babel command was:
./obabel.exe -:"c1c(ccc(c1)C(=O)OC)C#Cc2ccc(cc2)N(C)C" --gen3d -O rod.gzmatNote that this creates the molecule in 3D directly from the smiles string and relies on open babel’s default optimisation which may or may not be appropriate.
The atomic numbering is also different to the example above. It is (click to zoom):
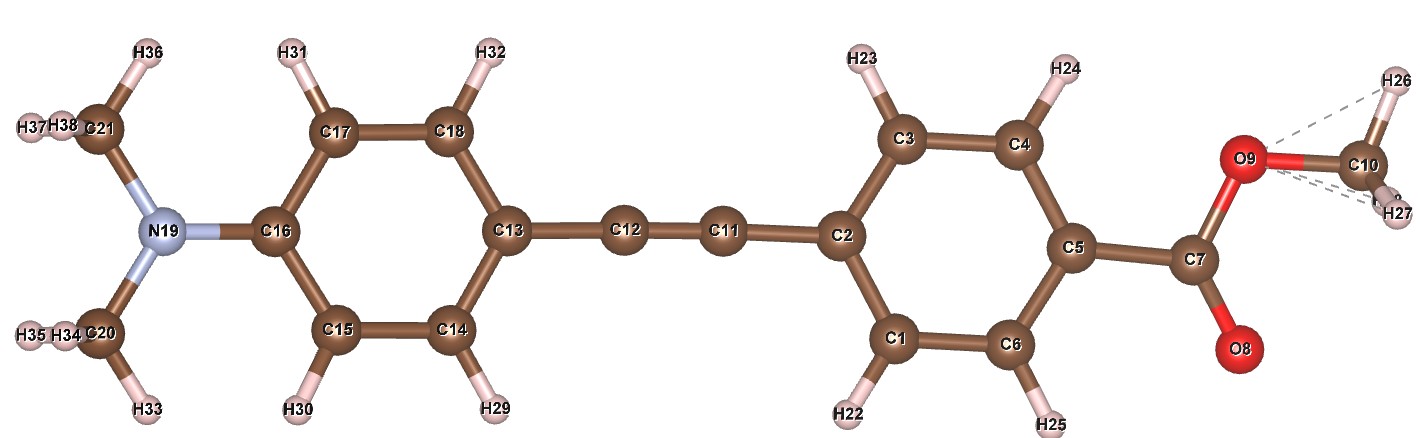
The Z-matrix is:
rigid
load z_matrix {
C1
C2 C1 1.3975
C3 C2 1.3992 C1 119.67
C4 C3 1.4021 C2 120.26 C1 0.57
C5 C4 1.4008 C3 119.97 C2 359.75
C6 C1 1.3975 C2 120.19 C3 359.79
C7 C5 1.4838 C4 122.60 C3 180.76
O8 C7 1.2208 C5 123.22 C4 153.43
O9 C7 1.3572 C5 110.73 C4 330.69
C10 O9 1.4302 C7 113.39 C5 183.00
C11 C2 1.4322 C1 119.49 C3 179.48
C12 C2 2.6376 C1 118.57 C3 178.78
C13 C2 4.0693 C1 117.85 C3 177.94
C14 C13 1.3920 C12 120.66 C11 58.54
C15 C14 1.4059 C13 120.34 C12 181.48
C16 C15 1.4052 C14 122.35 C13 0.16
C17 C16 1.4054 C15 115.80 C14 359.38
C18 C13 1.3909 C12 120.56 C11 239.52
N19 C16 1.3842 C15 122.23 C14 179.37
C20 N19 1.4586 C16 122.59 C15 28.22
C21 N19 1.4594 C16 122.36 C15 209.31
H22 C1 1.0875 C2 120.39 C3 179.97
H23 C3 1.0878 C2 120.23 C1 180.57
H24 C4 1.0875 C3 119.39 C2 180.47
H25 C6 1.0885 C1 119.52 C2 179.99
H26 C10 1.0935 O9 108.31 C7 178.26
H27 C10 1.0955 O9 110.83 C7 59.63
H28 C10 1.0946 O9 110.30 C7 296.83
H29 C14 1.0871 C13 120.43 C12 0.64
H30 C15 1.0864 C14 117.21 C13 177.87
H31 C17 1.0861 C16 120.39 C15 182.29
H32 C18 1.0868 C13 120.44 C12 358.37
H33 C20 1.0931 N19 114.44 C16 3.47
H34 C20 1.0955 N19 110.24 C16 241.05
H35 C20 1.0966 N19 109.31 C16 122.77
H36 C21 1.0932 N19 114.48 C16 1.84
H37 C21 1.0954 N19 110.08 C16 239.60
H38 C21 1.0965 N19 109.47 C16 121.28
}
macro rotcon {val_on_continue = Rand(-180,180);}
Rotate_about_axies(xrota -160 rotcon, yrota -40 rotcon, zrota 17 rotcon )
Translate(xtransa 0.15, ytransa -0.07, ztransa 0.87)4. If you read the .gzmat file directly it will create the necessary atomic sites. If not you can create them quickly in topas-editor. Hold down shift-alt and copy the site labels. Paste them above the Z-matrix and column edit so you have a series of lines as below. The coordinates can all be set to zero as they’ll be generated by the rigid body.
site C1 x 0 y 0 z 0 occ C 1 beq bvalue 1
site C2 x 0 y 0 z 0 occ C 1 beq bvalue 1
..
..
site H38 x 0 y 0 z 0 occ H 1 beq bvalue 15. Try a Rietveld refinement. You should get Rwp ~8.8 %.
6. The Z-matrix description above has sensible starting positions and angles for the molecule. If you didn’t know these you could determine them by simulated annealing. Try setting them all to zero, adding “continue_after_convergence” at the top of the file and refining. You should find the correct approximate structure is found within a few thousand cycles. At this point you’ve solved the structure based solely on the smiles 2D description.
7. The Z-matrix description from open babel has a significant torsion of the CO2 and NMe2 groups relative to the benzene rings. Try refining the relevant torsion angles by modifying the Z-matrix description as below. You should get Rwp ~7 %.
O8 C7 1.2208 C5 123.22 C4 tor_co2 169.58508`
O9 C7 1.3572 C5 110.73 C4 =tor_co2+180;:349.58508`
C20 N19 1.4586 C16 122.59 C15 tor_nme2 1.25098`
C21 N19 1.4594 C16 122.36 C15 =tor_nme2+180;:181.25098`8. The model here is reasonably close to the single crystal structure. You could try improving the fit by refining the torsion angle between benzene rings, refining some of the bond distances in the Z-matrix, etc.
9. In case you need it there is a cheat file here.