Solution
TOPAS-Academic – Tutorial
Structure solution of PbSO4
This tutorial uses the indexing results of the previous tutorial. The indexing results produced two possible space groups Pna21 and Pnma; for brevity we will use the correct one and in the right setting Pnam.
We will also introduce a number of techniques for speeding up the structure solution process.
Load the peak fitting INP file TA-Tutorials\data\pbso4-indexing\pbso4.inp. Save this file as pawley.inp.
Modify the file to perform a Pawley refinement for determining non-structural parameters accurately; the INP file should look like the following:
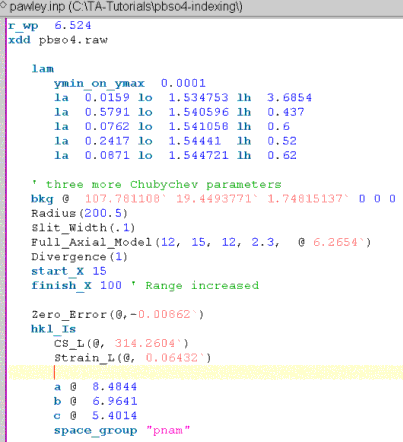
Note, a Zero_Error has been added using the XInsert node “Scan file level/Corrections”. The x-axis range has been increased to 100 degrees 2Th and three more Chubychev parameters have been added.
Save pawley.inp to a file called solution.inp. Modify this file for structure solution to look like:
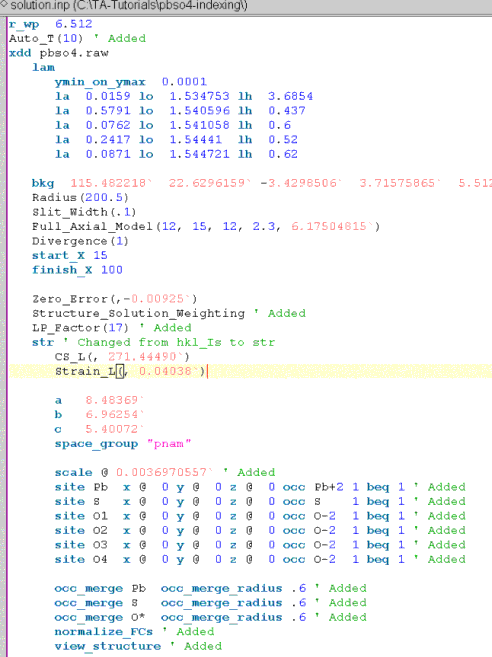
Refinement of the background have been turned off by removing the ‘@’ charcater after the bkg keyword.
The following have been added:
A Lorentz Polarization factor found under the the XInsert node “Scan file level/Corrections”.
Auto_T and Structure_Solution_Weighting macros, both found under the XInsert node called “Structure Solution”.
The Rietveld scale parameter “scale @ .001”.
normalize_FCs which normalizes the fractional coodinates to within 0 and 1 after refinement.
view_structure which displays an OpenGL window showing the structure in 3D.
Only the fractional atomic coordinates and the scale parameter are refined.
Typically the number of sites can be guessed to some accuracy from the volume of the unit cell. However the possibility of atoms residing on special positions complicates matters. To cater for special positions the occ_merge keywords have been included so that atoms of the same species approaching each other have their occupancies reduced (Favre-Nicolin, V. and Cerny, R. “Fox: Modular Approach to Crystal Structure Determination from Powder Diffraction”, EPDIC 8 proceedings, 2002.) , search the Technical Reference for occ_merge.
 in the Fit Window toolbar; this speeds up the structure solution process. Also consuming CPU time is the Rwp plot of the Fit Window; it can be turned off by displaying only “Text” in the fit window which is accomplished by clicking on the left most icon of the Fit Window toolbar.
in the Fit Window toolbar; this speeds up the structure solution process. Also consuming CPU time is the Rwp plot of the Fit Window; it can be turned off by displaying only “Text” in the fit window which is accomplished by clicking on the left most icon of the Fit Window toolbar.The simulated annealing process continues until the maximum number of iterations as defined by the keyword iters is reached, if undefined then iters defaults to 500000. Typically the User stops the simulated annealing process by clicking on the green arrow of the Fit Window.
Finding the correct solution takes less than 20 seconds on a 3GHz computer. To speed up the process the Decompose(0.005) macro, found under the XInsert node of “Structure Solution”), can be inserted at the scan level. The Decompose macro contains the keyword yobs_to_xo_posn_yobs, search for it in the Technical Reference.
The TA screen during structure solution and with Decompose looks like the following:
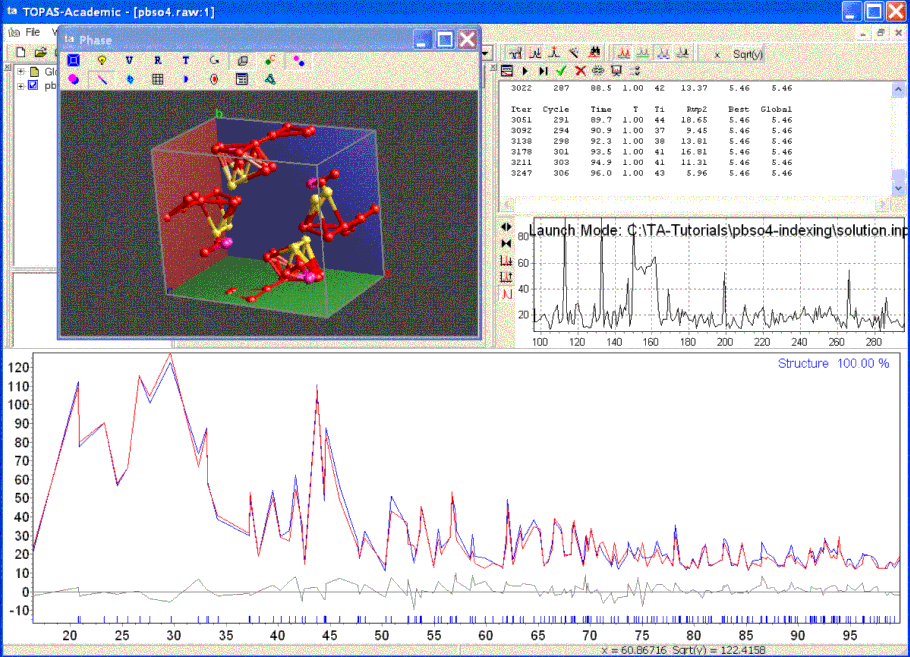
When the structure solution process is terminated by clicking on the green arrow the parameters that produced the best solution is saved and the OpenGL window and the calculated pattern is updated to reflect the best solution.
Inspection of the OpenGL window reveals which atoms lie on top of each other and thus special positions and degenerate sites can be easily identified. Atoms of the same site that lie on top of each other identifies a special position. Atoms of different sites that lie on top of each other identifies degenerate sites. In the case of this structure two oxygen sites merge into one with a corresponding occupancy of around 0.5. Similar deductions can be made by inspection of bond lengths as outputted using the append_bond_lengths keyword.